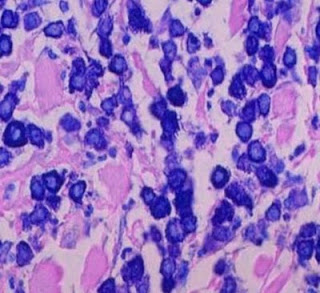
Answer of Dermatopathology Case 97

Immunohistochemistry findings: CD3 CD4 CD7 CD20 Mycosis Fungoides-associated Follicular Mucinosis: Abstract: Follicular mucinosis: a clinicopathologic, histochemical, immunohistochemical and molecular study comparing the primary benign form and the mycosis fungoides- associated follicular mucinosis.J Cutan Pathol. 2009 Jul 14. Objectives: To determine (i) whether primary (idiopathic) follicular mucinosis (PFM) and lymphoma-associated follicular mucinosis (LAFM) are distinct or related entities and whether there are reliable criteria that allow the two forms to be distinguished, (ii) the histochemical properties and consequently the type of mucin that accumulates in the follicle in PFM and LAFM, and (iii) whether there is any difference between the staining properties of mucin in patients with PFM and LAFM. Methods: Thirty-one patients were divided into two groups. Group 1 comprised 20 patients with no associated mycosis fungoides or Sézary syndrome (PFM) and group 2 was made up of