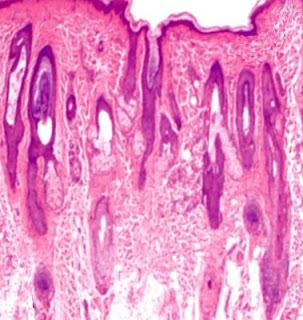
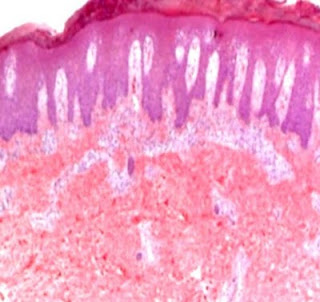
Answer of Dermatopathology Case 85

Graft-Versus-Host Disease Visit: Dermatopathology site Visit: Cutaneous Lesions in Graft versus Host Disease Abstract: Clinicopathologic characteristics of cutaneous chronic graft-versus-host diseases: a retrospective study in Korean patients. Int J Dermatol. 2010 Dec;49(12):1386-92. BACKGROUND: Chronic graft-versus-host disease (cGVHD) is a major complication in long-term survivors of hematopoietic stem cell transplantation (HSCT). Cutaneous manifestations are frequently the presenting features; therefore, the dermatologist needs to be aware of the wide spectrum of cutaneous cGVHD. METHODS: We retrospectively evaluated patients' characteristics, clinical, and histological features of cutaneous cGVHD and analyzed factors influencing the severity of cutaneous cGVHD in 100 Korean HSCT recipients between January 1, 1995, and December 31, 2007. RESULTS: Clinical manifestations of cutaneous cGVHD mainly presented as lichenoid (60.0%), sclerodermoid (12.0%), or erythematous maculopapul...