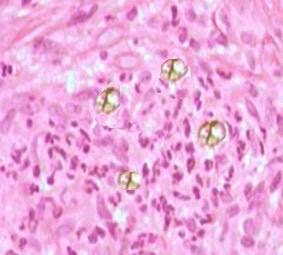
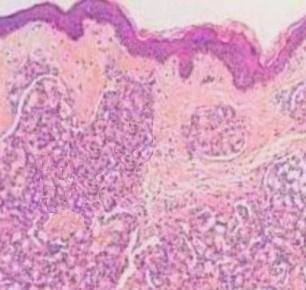
Answer of Dermatopathology Case 37
Dermatitis Herpetiformis Visit: Dermatopathology site Abstract: Dermatitis herpetiformis. An update of the pathogenesis.Hautarzt. 2009 Aug;60(8):627-30, 632. The multifactorial pathogenesis of dermatitis herpetiformis is reviewed in light of current experimental data. Genetic background, gluten consumption, and abnormal immune and autoimmune reactions are the most important pathogenetic factors, but other agents also participate in the disease development. The predisposing and inducing factors are summarized, while the pathophysiological steps leading to the development of skin symptoms are detailed. Dermatitis herpetiformis. A clinical chameleon.Hautarzt. 2006 Nov;57(11):1021-8; quiz 1029. Celiac disease is a genetically determined bowel disease also influenced by exogenous factors in which exposure to grain components triggers a chronic immune response with intestinal symptoms. Dermatitis herpetiformis represents the cutaneous manifestation of celiac disease. While intense pruri...