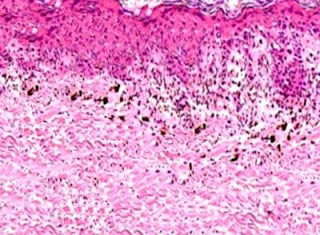
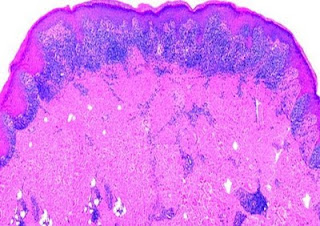
Answer of Dermatopathology Case 64

Immunohistochemistry: CD34 is strongly positive. Dermatofibrosarcoma Protuberans Visit: Dermatopathology site Abstract: Cutaneous CD34+ Spindle Cell Neoplasms: Histopathologic Features Distinguish Spindle Cell Lipoma, Solitary Fibrous Tumor, and Dermatofibrosarcoma Protuberans.Am J Dermatopathol. 2010 Jun 17. Spindle cell lipoma (SCL), dermatofibrosarcoma protuberans (DFSP), and solitary fibrous tumors (SFT) are cutaneous CD34+ spindle cell tumors that may exhibit histopathologic and immunophenotypic overlap. We sought ways to reliably distinguish among these lesions even in small or superficial biopsies. Ten morphologic characteristics were analyzed in a group of 5 SCLs, 6 cutaneous SFTs, and 12 DFSPs. SFT and DFSP exhibited extensive histopathologic overlap in small or partial biopsies. However, adnexal entrapment, defined as diffuse proliferation of tumor cells around pilosebaceous and eccrine structures with minimal disruption or expansion of the dermis, was a feature seen in 10 o...